


CONTACT
Bangalore, IndiaPhone: +91 80 2293 3109
Email: sudeep [at] chemeng.iisc.ernet.in
Openings in the group
WELCOME
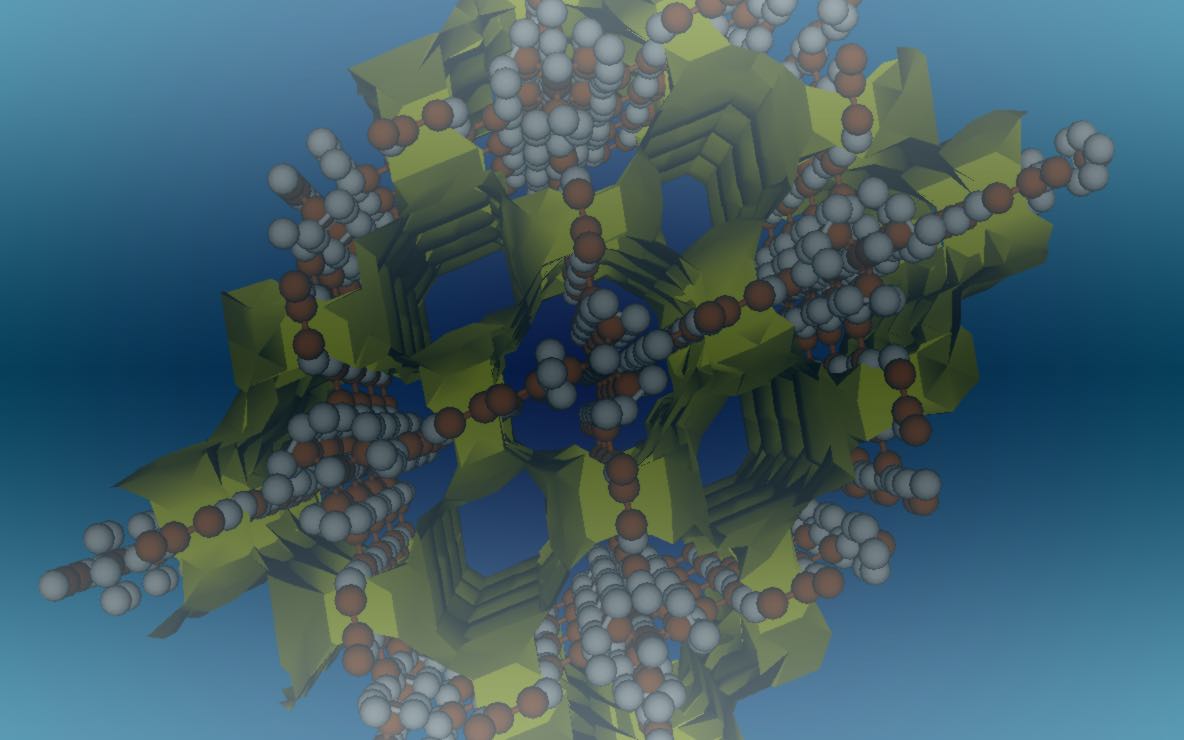
The research in our group is directed towards understanding the relationship between macroscopic thermodynamic behavior and microscopic interactions using the principles of statistical mechanics. The physical insights gained from such studies can help us in developing molecular theories to determine thermodynamic properties. They also aid in the development of molecular models to predict bulk behavior using molecular simulations. Our current focus is on understanding solid-fluid equilibrium in nanoparticle suspensions, thermodynamic behavior of gas-hydrate systems and thermodynamics of adsorption in microporous materials such as zeolites, metal-organic frameworks (MOFs), etc. The primary tools used in our research include molecular simulations ( both Monte Carlo and molecular dynamics) and density functional theories.