Researchers at the Chemical Engineering at IISc and Unilever have developed advanced computational models of bacterial cell walls to accelerate the screening of antimicrobial molecules. These models simulate the intricate structures of Gram-negative bacteria like E. coli and Gram-positive bacteria like S. aureus, providing a detailed understanding of how antimicrobials interact with these cellular barriers. K G Ayappa Lab’s atomistic model for S. aureus, which captures the cell wall’s structure at an atomic level, revealed how antimicrobials such as melittin and thymol disrupt bacterial cell walls by interacting with key peptides and translocating through peptidoglycan layers.
Another study focused on the behavior of surfactant molecules moving through the E. coli cell wall. The researchers discovered that shorter surfactant chains, like laurate, translocated more efficiently than longer chains, such as oleate, which correlated with higher bacterial killing rates. This insight was validated by experiments showing that shorter chain surfactants caused bacterial vesicles to burst more rapidly. The overarching aim of this collaboration is to utilize these computational models for the rapid screening of potential antimicrobials, streamlining the process of identifying promising candidates for laboratory testing.
References:
Sharma P, Vaiwala R, Parthasarathi S, Patil N, Verma A, Waskar M, Raut JS, Basu JK, Ayappa KG, Interactions of surfactants with the bacterial cell wall and inner membrane: Revealing the link between aggregation and antimicrobial activity, Langmuir (2022)
Vaiwala R, Sharma P, Ayappa KG, Differentiating interactions of antimicrobials with Gram-negative and Gram-positive bacterial cell walls using molecular dynamics simulations, Biointerphases (2022)

Paper has found an important role in conducting bioanalytical reactions in low resource settings, where expensive analytical instruments may be lacking. The Whitesides group at Harvard University first introduced microfluidic paper analytical devices (μ PADs) as low-cost paper devices for conducting multiplexed colorimetric detection of analytes in a fluid. Since then, hundreds of groups around the world have developed μPADs for the detection of various analytes. The fabrication of μPADs requires patterning of flow barriers using wax or other hydrophobic materials to create flow channels in paper, which is a bottleneck in large scale fabrication. Bhushan Toley’s group has developed a new barrier-free design in which multiplex colorimetric detection is accomplished, without having to pattern any barriers in paper. Barrier-free μPAD (BF μPAD) was accomplished by a paper stacking strategy in which multiple detection reagent spots are deposited on the bottom layer and the sample fluid is introduced into the top layer. The strategic combination of paper layers is such that it eliminates lateral convection in the bottom membrane. As a result, the multiple test zones in the bottom membrane do not merge into each other. Fabrication of multiplex colorimetric detection paper-based devices simply by stacking paper membranes may enable massive scaleup of such devices in the near future.
Reference: Ayushi Chauhan and Bhushan J. Toley, Barrier-Free Microfluidic Paper Analytical Devices for Multiplex Colorimetric Detection of Analytes, Anal. Chem. 2021, 93, 25, 8954–8961

A quarter of the world’s population is expected to develop urinary tract infection (UTI) at least once in their lifetime. Currently, patients that walk into the doctor’s clinic with UTI-like symptoms are often prescribed antibiotics empirically. The standard test that determines the kind of infecting pathogen and its resistance status to various antibiotics takes 2-3 days to complete. Therefore, the clinician does not have enough evidence of whether the antibiotic they are prescribing will treat the infection. Neither do they have rapid tools at their disposal to get this information. Consequently, antibiotics are prescribed empirically. In case, the causative organism is resistant to the prescribed antibiotic, the prescribed antibiotic will be unsuccessful in treating the infection and may lead to further mutation of the organism making it more resistant to the administered antibiotic. This practice of empirical administration of antibiotics is leading to an increasing burden of Anti-Microbial Resistance (AMR) globally. The world needs disruptive tools that can enable the clinician to determine drug sensitivity rapidly, preferably during the patient’s visit to the clinic. Papyrus Diagnostics Pvt. Ltd., a company founded by Prof. Bhushan Toley, has ideated simple paper-based devices that may enable rapid AMR testing of UTIs in under an hour. Papyrus’s proposal presented at the C-CAMP AMR Idea Diagnostics Challenge 2022 was the 1st runner-up in this national idea challenge, which saw more than 100 ideas from across the country.
COVID-19 vaccines have been a game-changer in the current pandemic. Several vaccine candidates have conferred a high degree of protection, with some reducing symptomatic infections by over 95%. What determines this extent of protection remained poorly understood.
In a recent study, Narendra Dixit, a professor in our department, together with alumni Pranesh Padmanabhan and Rajat Desikan, developed a mathematical model that predicts how antibodies generated by COVID-19 vaccines confer protection against symptomatic infections. This would help not only understand the workings of COVID-19 vaccines but also optimise their use and speed up the development of new ones.
Predicting vaccine efficacies has been hard because the processes involved are complex and operate at many interconnected levels. Vaccines trigger different antibodies, each affecting virus growth differently. This in turn affects the dynamics of the infection and the severity of symptoms. Further, different individuals generate different collections of antibodies and in different amounts. To address these challenges, the authors analysed over 80 different neutralising antibodies generated after vaccination. They hypothesised that these antibodies constitute a ‘landscape’ and that each individual produces a unique ‘profile’ of antibodies that is a small, random subset of this landscape. They then developed a mathematical model to simulate infections in a virtual patient population of many thousand people with different antibody profiles. Model predictions closely matched efficacies reported in clinical trials for all the major approved vaccines. Further, the model showed that vaccine efficacy was linked to a readily measurable metric called antibody neutralization titre, opening up the possibility of using the model to test future vaccines for their efficacies before elaborate clinical trials are launched.
Reference:
Padmanabhan P, Desikan R & Dixit NM. Modeling how antibody responses may determine the efficacy of COVID-19 vaccines. Nature Computational Science (2022)

Our body is home to a large number of microbes. Our health depends on them. For instance, disruption of the microbial communities in our gut, which can happen with the use of antibiotics, affects not only our digestion but also our mental health. Microbial communities are also found in nearly every environmental niche, including the soil and the oceans, and are central players in the respective ecosystems. More recently, recognizing their power, artificial microbial communities are being assembled for numerous applications, such as biofuel production. It is important, therefore, to understand how microbial communities thrive and devise ways of engineering them.
In a recent study, Prof. Narendra Dixit together with his M. Tech. student Aamir Ansari developed a new method to efficiently predict the compositions of microbial communities. The study was performed in collaboration with Unilever R&D in Bengaluru. The method, termed EPICS, involves the use of effective pairwise interactions to predict community structures.
A major challenge in such prediction was the large number of interactions between the microbes present, which determine the compositions of microbial communities but remain difficult to unravel. In the new method, the researchers developed a way to subsume higher order interactions into effective pairwise interactions, which were easier to unravel and, at the same time, facilitated accurate prediction of community compositions. The method dramatically reduces the effort in unravelling the interactions and enables the study of much larger microbial communities than currently feasible. They demonstrated its applicability using a synthetic oral microbiome. The method brings us a step closer to understanding the microbiomes pervading our bodies and surroundings and developing microbiome-based interventions.
Reference:
Ansari AF, Reddy YBS, Raut J, Dixit NM. An efficient and scalable top-down method for predicting structures of microbial communities, Nature Computational Science (2021)
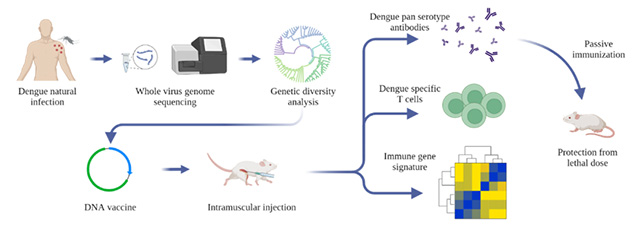
Dengue virus (DENV) is a mosquito-borne flavivirus with a single-stranded RNA genome that causes an estimated 390 million infections worldwide every year. All four serotypes of DENV (DENV1-4 sharing 65-70% sequence homology) are co-circulating in India simultaneously. Although more than a hundred thousand dengue cases are reported from India every year, the genomic diversity of the prevalent dengue viruses (DENVs) in the country is largely unknown. Using whole-genome sequencing of viral RNA from clinical human serum samples, Rahul Roy’s group report 119 DENV full-genomes from four metropolitan sites across India from 2012 to 2018.
It has been known that homotypic secondary infection is generally asymptomatic, but heterotypic secondary infection is associated with increased chances of severe infection. The cross-reactive antibodies can enhance subsequent infection by a heterologous serotype (ADE: antibody-dependent enhancement). The envelope protein domain III (EDIII) has been identified as the major target of neutralizing and serotype-specific antibodies, while precursor membrane and EDI-II-directed antibodies are reported to enhance infection via ADE. Recent studies have found that EDIII-based DENV vaccines could circumvent ADE of infection in mice. Another target for the vaccine is NS1, which is a highly conserved protein among flaviviruses and has been shown to be evoking antibody and T cell responses previously.
Team of researchers from NCBS, C-CAMP and IISc developed a DENV EDIII-based DNA vaccine candidate by integrating the consensus sequences from the circulating sequences of each serotype, incorporating the NS1 protein-coding region of DENV2. The vaccine generated robust antibody response and T cell response against all the four serotypes of DENV. Passive transfer of antibodies from the immunized mice also protected the non-immunized mice from the lethal dose of DENV infection.
Reference:
A Sankaradoss, S Jagtap, et al., Immune profile and responses of a novel Dengue DNA vaccine encoding EDIII-NS1 consensus design based on Indo-African sequences, Molecular Therapy (2022)