Researchers at the Chemical Engineering at IISc and Unilever have developed advanced computational models of bacterial cell walls to accelerate the screening of antimicrobial molecules. These models simulate the intricate structures of Gram-negative bacteria like E. coli and Gram-positive bacteria like S. aureus, providing a detailed understanding of how antimicrobials interact with these cellular barriers. K G Ayappa Lab’s atomistic model for S. aureus, which captures the cell wall’s structure at an atomic level, revealed how antimicrobials such as melittin and thymol disrupt bacterial cell walls by interacting with key peptides and translocating through peptidoglycan layers.
Another study focused on the behavior of surfactant molecules moving through the E. coli cell wall. The researchers discovered that shorter surfactant chains, like laurate, translocated more efficiently than longer chains, such as oleate, which correlated with higher bacterial killing rates. This insight was validated by experiments showing that shorter chain surfactants caused bacterial vesicles to burst more rapidly. The overarching aim of this collaboration is to utilize these computational models for the rapid screening of potential antimicrobials, streamlining the process of identifying promising candidates for laboratory testing.
References:
Sharma P, Vaiwala R, Parthasarathi S, Patil N, Verma A, Waskar M, Raut JS, Basu JK, Ayappa KG, Interactions of surfactants with the bacterial cell wall and inner membrane: Revealing the link between aggregation and antimicrobial activity, Langmuir (2022)
Vaiwala R, Sharma P, Ayappa KG, Differentiating interactions of antimicrobials with Gram-negative and Gram-positive bacterial cell walls using molecular dynamics simulations, Biointerphases (2022)

Although seemingly simple, the nucleation of crystals from the fluid phase is a complex phenomenon. In spite of numerous computational and experimental studies, its mechanism is not completely understood. Recent studies have shown that, contrary to century-old classical nucleation theory, crystal nucleation follows a two-step mechanism where the molecules initially come together to form an aggregate, followed by crystal nucleation within the aggregate. In chemical process industries, an understanding of the nucleation mechanism is important for design and control of crystallization processes. For example, in the pharmaceutical industry, polymorph control (control of crystal structure) during crystallization is of vital technological and commercial importance.
In such a scenario, Ravi Kumar Reddy Addula and Sudeep N Punnathanam from the Department of Chemical Engineering have developed a new molecular theory of crystal nucleation theory from dilute phases such as vapours and dilute solutions. The molecular theory with its basis in statistical mechanics, is able to provide the most complete description till date of the nucleation process and is expected to provide important insights into the mechanism of crystal nucleation from solutions. This should enable scientists and engineers to make improvements in process design and control of crystallization process.
Reference:
Ravi Kumar Reddy Addula and Sudeep N. Punnathanam, Molecular Theory of Nucleation from Dilute Phases: Formulation and Application to Lennard-Jones Vapor, Phys. Rev. Lett. (2021)
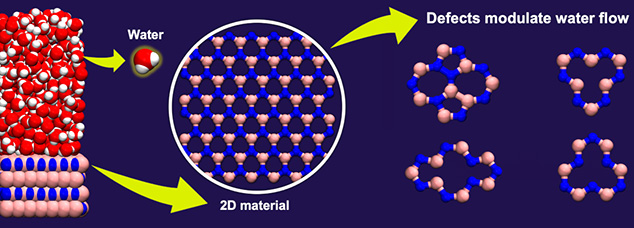
Two-dimensional (2D) materials, which consist of a few layers of atoms, are currently being explored for various applications, including seawater desalination, oil-water separation, and osmotic power harvesting. These materials often contain imperfections or defects in them, which can alter the properties of the material. Although defects in materials are usually thought to increase frictional forces, computer simulations show a notably different picture. These unexpected results were published in the journal Nano Letters by Ananth Govind Rajan, an Assistant Professor in our department, and Aniruddha Seal, an undergraduate student from the National Institute of Science Education and Research.
The team combined quantum-mechanical calculations of the charge distribution in hexagonal boron nitride (hBN), a 2D material, with classical molecular dynamics simulations of water interacting with hBN. Molecular dynamics simulations allow researchers to model the trajectories of atoms and molecules and to make predictions regarding frictional behaviour at the nanoscale.
Surprisingly, the authors found that certain defects, such as the nitrogen (N) vacancy (where a nitrogen atom is missing from the hBN lattice) and the Stone-Wales defect (where a boron-nitrogen bond is rotated by 90 degrees in the hBN lattice), led to a reduction in water-hBN friction. On the contrary, a boron (B) vacancy increased water-hBN friction almost threefold. The authors quantified these predictions in terms of the “slip length”, a quantity that determines to what extent water molecules “slip” on the hBN surface. They found that, at high defect concentrations, the slip length of hBN with a N vacancy could be comparable to that of graphene, the most slippery 2D material known to date. These findings have implications for the optimal and predictive design of devices made using hBN.
Reference: Seal, A.; Govind Rajan, A. Modulating Water Slip Using Atomic-Scale Defects: Friction on Realistic Hexagonal Boron Nitride Surfaces. Nano Lett. 2021, 21, 19, 8008-8016

Researchers at IISc and EPFL have developed a breakthrough in water evaporation technology using nanoporous graphene with pores on the angstrom scale. This innovative approach increases the water evaporation rate by 35 times compared to traditional methods. The key lies in the confinement of water molecules within these tiny pores, which reduces hydrogen bonding at the edges, making it easier for the molecules to escape as vapor.
In the new study, Anshaj Ronghe and Prof. K Ganapathy Ayappa from the Department of Chemical Engineering (CE), IISc and collaborators have developed a novel, low thermal input, evaporation-based process using single layered graphene sheets synthesised with tunable-sized nanopores. Enhancing the kinetics of liquid–vapor transition from nanoscale confinements is an attractive strategy for developing evaporation and separation applications. The ultimate confinement limit for evaporation is an atom-thick interface hosting angstrom-scale nanopores. This advancement could revolutionize water purification and desalination processes, offering a highly efficient and low-energy solution to provide clean water in regions facing water scarcity.
Reference:
Lee WC, Ronghe A, et al., Enhanced Water Evaporation from Å-Scale Graphene Nanopores, ACS Nano (2022)