A variety of developments such as faster computers, realistic force-fields and sophisticated algorithms, have spurred the use of statistical mechanics and molecular simulations towards such research. The Supercomputer Education and Research Centre (SERC) at IISc houses the CRAY XC40 which provides a 1 petaflop computing facility serving the entire campus community. In our department, faculty have been using these tools for research in a wide variety of areas.
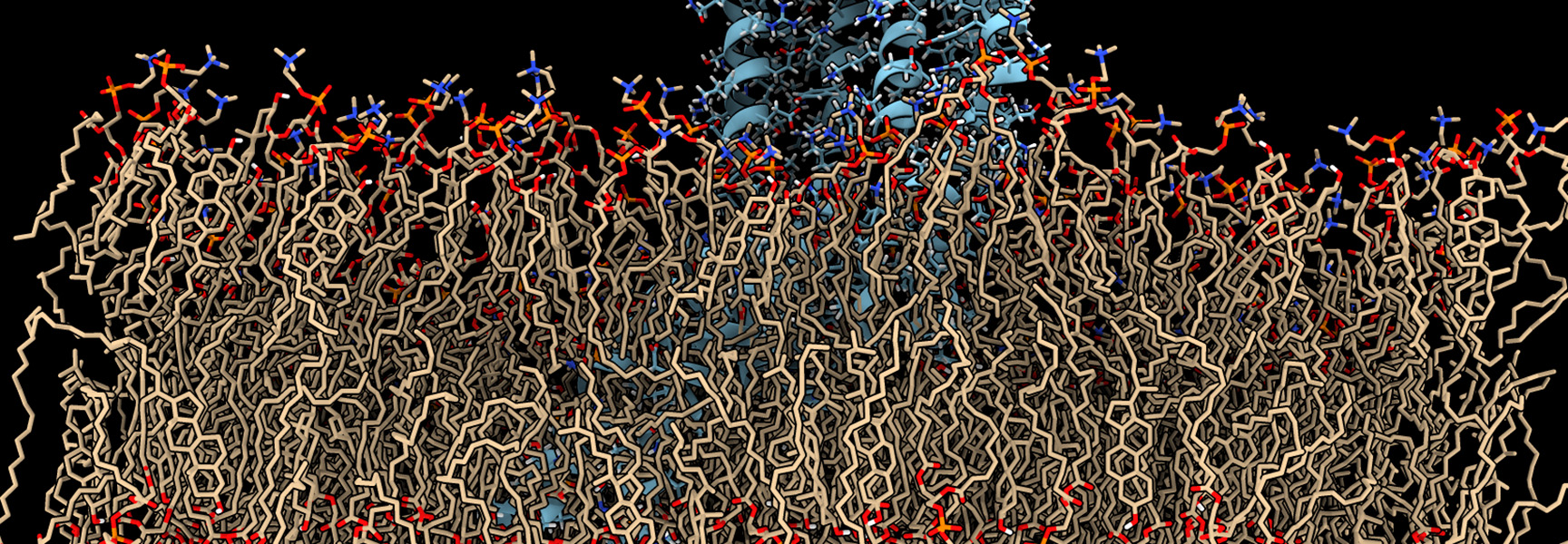
Atomistic molecular dynamics simulations of the lamellar phase are used to extract structural and mechanical properties which are input into large scale Lattice Boltzmann simulations to unravel the rheology of these complex mesoscale systems and connect macroscopic rheology with molecular constituents. Ayappa and co-workers have used atomistic as well as coarse grained molecular dynamics simulations to study the interaction of pore forming toxins with lipid bilayers. Micro-second long atomistic simulations of systems with over half a million atoms have been used to reveal interactions of proteins with membrane cholesterol, stability of partially formed oligomers, dynamics of lipid molecules and mechanical properties of membranes.